PRESS RELEASE (技術)
2018年5月7日
株式会社富士通研究所
富士通株式会社
新薬候補を効果的に創出する分子シミュレーション技術を開発
薬効の目安となるパラメーターの推定誤差を10分の1以下に
株式会社富士通研究所(注1)(以下、富士通研究所)は、創薬向けの技術として、疾病の原因となるタンパク質(以下、標的タンパク質)と薬の候補となる化学物質が引き合う強さである結合強度を精度よく推定できる分子シミュレーション技術を開発しました。
創薬の過程において、標的タンパク質と化学物質の結合強度は、薬効の目安となることから正確に予測することが求められています。従来、結合強度を予測する方法として、分子内の原子間に働く力をニュートン力学により近似的に算出する分子シミュレーション技術が広く用いられてきましたが、最も重要なパラメーターである結合部分のねじれ度合いの推定精度が低いため、結合強度の推定精度も低くなってしまうことが課題でした。
今回、結合強度の予測値に直結する、化学物質のねじれ度合いについて、ねじれが発生する結合部分だけでなくその近傍の原子の影響まで考慮して推定する、分子シミュレーション技術を開発しました。
190種類の化学物質で、第一原理計算(注2)による正しい結果との誤差を評価したところ、従来技術に比べてねじれ度合いの推定誤差が平均で10分の1以下であることが確認できました。これにより、標的タンパク質と化学物質の結合強度を精度よく推定できるため、IT創薬へ活用することで従来の知見からでは得られない画期的な新薬創出が期待できます。
今後、富士通株式会社が提供する新たなIT創薬サービスにおいて、本技術を搭載する予定です。

新薬候補物質が標的タンパク質に結合する様子
(再生時間: 23秒 / 音声なし)
開発の背景
新薬の開発には十数年にわたる期間と多額の費用が必要なため、新たな新薬開発の手法が世界的に模索されており、その中で、コンピュータを利用してこれまでにない薬の候補となる化学物質を高い確率で創出可能な新薬開発手法(IT創薬)に高い関心が寄せられています。IT創薬により、これまでの化学物質の生成と実験を繰り返す試行錯誤での方法ではなく、化学物質を仮想的に設計し、効果を見積もることが可能となるため、新薬を創出する画期的な技術として期待されています。
課題
化学物質の薬としての効果は、化学物質が標的タンパク質に結合することで発現します。化学物質は標的タンパク質に結合する際に、標的タンパク質の形状に合わせて、形状を変化させます。この形状変化の程度を表すパラメーターとなる変形度合いは薬効の目安となる両者の結合強度に直結するため、正確に予測することが要求されます。
化学物質の変形度合いを算出する方法として、量子力学に基づく方法と、ニュートン力学を用いる方法がありますが、量子力学に基づいて原子の種類や位置から電子の状態を解く、第一原理計算は、厳密な算出が可能な反面、算出のために膨大な時間を要し、多数の化学物質の変形度合いをシミュレートするためには、年単位の時間を必要とするため、現実的ではありません。
一方で、分子内の原子間に働く力を、ニュートン力学を用い分子シミュレーションにより近似的に算出する方法は、高速であり標的タンパク質のような大きな分子も容易に取り扱えるため、広く用いられてきました。ニュートン力学では、原子間に働く力を、結合する2つの原子の距離に依存する力、結合する3つの原子の角度に依存する力、結合部分のねじれ度合いに依存する力、および結合していない原子間の距離に依存する力で表現しますが、中でも標的タンパク質と化学物質が結合する上では、結合部分のねじれ度合いが重要な変化度合いとなります。しかし、従来技術では、結合部分のねじれ度合いを算出するための二面角パラメーター(図1)の推定精度が低く、結果として、シミュレーションによる結合強度の推定精度が低いという問題がありました。
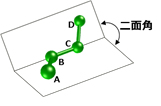
図1 二面角(原子A, B, Cの作る平面と、原子B, C, Dの作る平面が成す角)
開発した技術
富士通研究所は、10年以上にわたり分子シミュレーション技術の開発を行ってきました。今回、これまでの開発で培ってきた知見を活かし、結合部分の近傍の原子の影響を考慮して二面角パラメーターを推定する、分子シミュレーション技術を開発しました。
従来技術では、結合する2つの原子とそれぞれの原子に結合する原子の合計4つの原子に基づいて二面角パラメーターを推定しますが、分子の構造によっては、それ以外の原子の影響が大きくなることがあり、その場合に推定誤差が大きくなっていました。
今回、結合部分以外の原子の影響が大きくなるパターンの部分構造とその場合に推定される化学物質のねじれ度合いについて、推定式をデータベースとして整備しました。これにより、データベースに該当する分子の部分構造の場合にも、対応する推定式にてねじれ度合いを求めることで、従来は正確な算出が難しかった分子のねじれ度合いでも高精度な推定を可能としました(図2)。
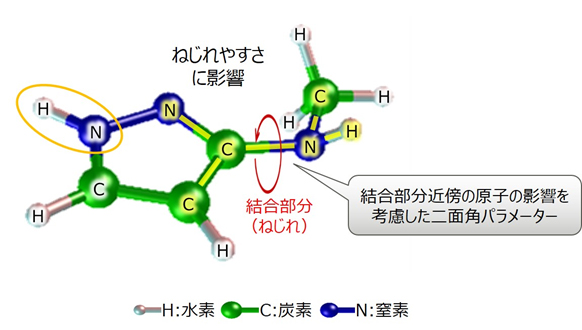
図2 分子構造の例:3-(メチルアミノ)ピラゾールの場合
本技術を、富士通研究所が開発した原子間に働く力の精緻なパラメーターを生成するソフトウェア(FF-FOM)に実装した結果、正確な計算と一致することを確認しました。
効果
190種類の化学物質で、ねじれ度合いの推定値について第一原理計算による結果との誤差を評価したところ、従来技術(注3)に比べて平均で10分の1以下となり、1molあたり0.6kcal以下と室温の熱揺らぎエネルギーを下回り、実用的であることが確認できました。これにより、標的タンパク質と化学物質の結合強度を精度よく推定できるため、IT創薬へ活用することで画期的な新薬創出が期待できます。
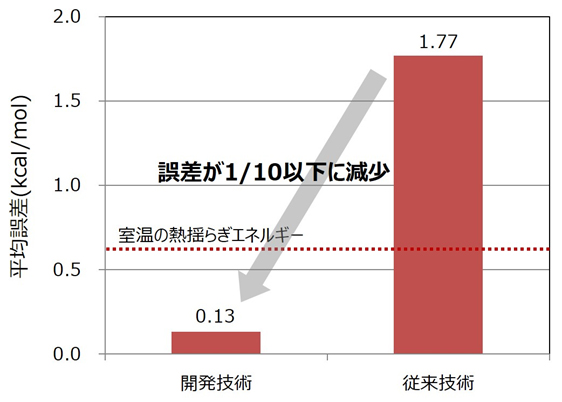
図3 二面角パラメーター値の性能評価
今後
富士通株式会社が今後提供予定のIT創薬に関するサービスに本技術を実装する予定です。
商標について
記載されている製品名などの固有名詞は、各社の商標または登録商標です。
以上
注釈
- 注1 株式会社富士通研究所:
- 本社 神奈川県川崎市、代表取締役社長 佐々木繁。
- 注2 第一原理計算:
- 量子力学に立脚した電子状態理論を用いた化合物などの性質を予測するシミュレーション手法。経験的なパラメーターを用いることなく、物性値を高精度に予測可能。他の計算手法と比べて計算負荷が非常に大きい。
- 注3 従来技術:
- デファクトスタンダードな位置エネルギーパラメーターセットGeneral AMBER Force Field 1.8(GAFF1.8)を使用。
本件に関するお問い合わせ
株式会社富士通研究所
デジタルアニーラプロジェクト
![]() 046-250-8376(直通)
046-250-8376(直通)
![]() fffom-pr@ml.labs.fujitsu.com
fffom-pr@ml.labs.fujitsu.com
プレスリリースに記載された製品の価格、仕様、サービス内容、お問い合わせ先などは、発表日現在のものです。その後予告なしに変更されることがあります。あらかじめご了承ください。